|
|
|
 |
 |
The usual usage of the grid architecture is running one computation on many distributed CPUs through a rapid network. However, in order to analyze much more complicated
biological systems, composed of simulations at different levels, along the new paradigm for biological science, more integrated computational approaches are required. The individual programs should be driven on their own corresponding machines on the grid system. For this purpose, we have designed and developed a new platform, BioPfuga (Biosimulation Platform United on Grid Architecture) where individual applications, corresponding to the different levels of bio-simulations, are united and executed as a hybrid application.
BioPfuga requires that (1) application programs are divided into a set of many pieces,
each of which corresponds to a unit simulation procedure, and that (2) data communication is made between the program pieces by a standard description.
For the former requirement, the simulation unit should not be too small for rapid computation, so that the data communication time among different machines is
minimal. For the latter problem, we have proposed a simple, standard description using XML, UDS-XML (Universal Data Set-XML) for the data exchange between different
computational programs for actual execution of Grid computations. Until now, many application programs used only binary data for intermediate and temporary
data in the field of High-Performance Computing (HPC). This was BioPfuga data files become very large and access very slow if data is described in a text format.
When the XML description is used, we designed three forms: a text form, a hexadecimal
form, and a Base64 form. In particular when the Base64 form is used, the size is only about 1.3 times larger than that used in the binary
form. The advantage of the XML form for intermediate and output data is that
any meta-data can be easily added as an attribute or as tagged information in
addition to the actual computed data to be exchanged among the different application
programs. It should be emphasized that the unit of data can always be provided
in UDS-XML, so that the different application programs recognize and confirm
the unit system for computation and analysis. Researchers in different scientific
fields usually use their own conventional units, and a barrier for research integration
is encountered without a common understanding of the units.
The schema of UDS-XML, examples for three forms (a text form, a hexadecimal form,
and a Base64 form), and the corresponding program library are provided at http://www.biogrid.jp/BioPfuga/uds-xml/.
People who are interested in BioPfuga, please request to the authors. |
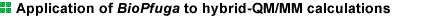 |
The static and dynamic features of protein structures are now analyzed by molecular
simulations. Chemical reactions at the active sites of proteins require information
about dynamic features of electrons, which cannot be attained by a classical
molecular mechanics simulation. Thus, we combine quantum chemical (QM) simulations,
AMOSS1), with the molecular mechanic (MM) simulations, prestoX-basic2, 3),
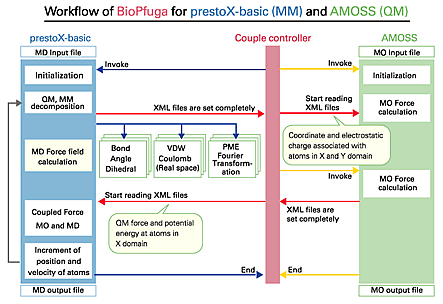
in a way that is suitable for Grid computation. We first divided the two big
programs into a set of many pieces. Then, a hybrid calculation was performed
following the work flow in Fig. 1. As a computational platform, we used PC
clusters composed of Pentium-3 and Pentium-4 processors, and the MD part was
also driven on the special-purpose computer for MD simulations, MDGrape24).
|
|
| Figure 1: |
Work flow of BioPfuga for a hybrid-QM/MM calculation using AMOSS1) and prestoX-basic2, 3).
|
As an example of this hybrid-QM/MM calculation, a simple model system composed
of ethanol in water was simulated (Fig. 2). Here, the ethanol molecule and
two water molecules that are close to the ethanol hydroxyl group are treated
as the QM region, and the Hartree-Fock molecular orbitals (MO) were analyzed
using 35 basis functions of MINI-45). The other 226 water molecules were represented
using a conventional classical model, TIP4P6). The Nose-Hoover algorithm was
applied at constant (283 K) temperature without any truncation of the non-bonded
interactions. The CAP boundary with the 13Á radius was used.
|
|
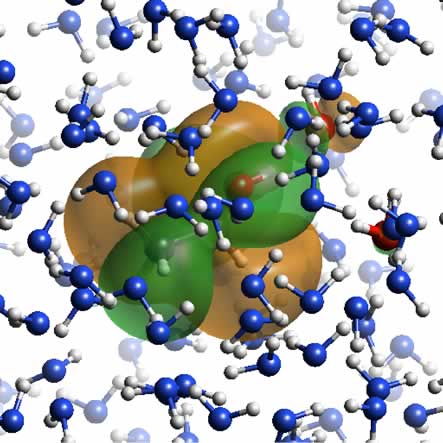
Figure 2:
|
A snap shot of the hybrid-QM/MM calculation for a simple model system composed
of ethanol in water.
The shape of the highest occupied molecular orbital
(HOMO) of the ethanol molecule and two water molecules close to the ethanol
hydroxyl group is shown by green (+0.003 e/au3) and orange (-0.003 e/au3).
The ball-and-stick models of the ethanol and the two water molecules are
also shown with the red oxygen atoms. The other 226 water molecules treated
by a classical TIP4P model are shown with the blue oxygen and white hydrogen
atoms.
|
From the canonical ensemble of the molecular system, it was found that the gauche
rotamer was as stable as the trans rotamer associated with the ethanol dihedral
angle around the -C-O- covalent bond. In contrast, when the classical force
field7) was used for ethanol, only the trans conformer was stable. When the
more precise basis functions are applied to ethanol, the more reliable results
can be obtained, and the force-field problems may be overcome. In the current
computational system, we used a Gigabit Ethernet among the PCs, and it took
about 0.1 s for 100 kBytes data transfer with the hexadecimal UDS-XML form
plus the corresponding XML parsing and writing procedures at one MD unit step.
|
 |
We propose BioPfuga as a new platform to integrate several different simulation programs for complicated analyses in bioscience fields. However, it is still preliminary leaving several unresolved problems.
First, the format of UDS-XML should be improved for more general and efficient use, not only for the hybrid-QM/MM calculations, but also for other integrated simulations covering continuum models for molecular systems8), and cell and organ simulations9). More semantic information should be added to the current form, with the aid of recent technology of the semantic Web. Second, the procedure for handling the XML data now requires longer CPU time than the corresponding procedure using the simple binary form. The XML interface should be revised for more speedy procedures. Third, the current Globus has no tools for the function of dynamic spawning, and so we mainly used MPI/LAM10) in the current implementation of BioPfuga. Fourth, for the integration between the Computing Grid and the Data Grid, all the files used in the simulation calculations should use the XML form, based on the expected rapid development of XML-Database systems. Finally, a new application for managing the work-flow using a GUI is also necessary for future advanced usage of BioPfuga.
These issues are now being overcome, with collaborative studies among the members of BioGrid in the groups of the core grid, data grid, and computing grid technologies.
|
 |
(1) Sakuma, T., Kashiwagi, H., Takada, T., and H. Nakamura, "Ab initio MO study of the chlorophyll dimer in the photosynthetic reaction center. I. A. theoretical treatment of the electrostatic field created by the surrounding proteins," Int. J. Quant. Chem., 61, 1, pp.137-151, 1997.
(2) Fukunishi, Y., Mikami, Nakamura, H., "The filling potential method: a method for estimating the free energy surface for protein-ligand docking," J. Phys. Chem. B, 2003, in press.
(3) Nakajima, N., Higo, J., Kidera, A., and Nakamura, H., "Free energy landscapes of peptides by enhanced conformational sampling," J. Mol. Biol., 296, 1, pp. 197-216, 2000.
(4) Narumi, T., Susukita, R., Ebisuzaki, T., McNiven, G., and Elmegreen, B., "Molecular dynamics machine: Special-purpose computer for molecular dynamics simulations," Mol. Simulation, 21, 5/6, pp. 401-415, 1999.
(5) Huzinaga, S., Gaussian Basis Sets for Molecular Calculations, Elsevier, New York, 1984.
(6) Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., "Comparison of simple potential functions for simulating liquid water," J. Chem. Phys., 79, 2, pp. 926-035, 1983.
(7) Jorgensen, W.L., "Optimized Intermolecular Potential Functions for Liquid Alcohols," J. Phys. Chem., 90, 7, pp. 1276-1284, 1986.
(8) Nakamura, H., "Roles of electrostatic interactions in proteins," Quart. Rev. Biophys., 29, 1, pp. 1-90, 1996.
(9) Noble, D., "Modeling the heart: from genes to cells to the whole organ," Science, 295, 5560, pp. 1678-1682, 2002.
(10) Squyres, J. M., Lumsdaine, A., George, W. L., Hagedorn, J. G., and Devaney, J. E., "The Interoperable Message Passing Interface (IMPI) Extensions to LAM/MPI," in Proceedings of MPIDC2000, 2000.
|
|
|